
EN

据统计,截止到2023年12月,全球已注册登记的干细胞相关临床项目多达4000余项,其中包含3101项已完成的项目,主要涉及自身免疫性疾病(如系统性红斑狼疮)、代谢性疾病(如糖尿病)、呼吸系统疾病(如慢阻肺)、骨关节病(如骨关节炎、类风湿关节炎)、生殖系统疾病(如男性勃起功能障碍)、肝脏疾病、眼科疾病等多个领域。
这其中,我国已有50余项间充质干细胞药物临床试验,包括糖尿病足溃疡、骨关节炎、类风湿关节炎、肺纤维化、卒中、银屑病、急性呼吸窘迫综合征、肝衰竭、克罗恩病、移植物抗宿主病等病种。随着更多临床试验数量增加,预计到2028年,中国干细胞市场经济的复合年均增长率将达到16.5%。
PART1 备案及上市的干细胞药物汇总
全球已上市的间充质干细胞药物
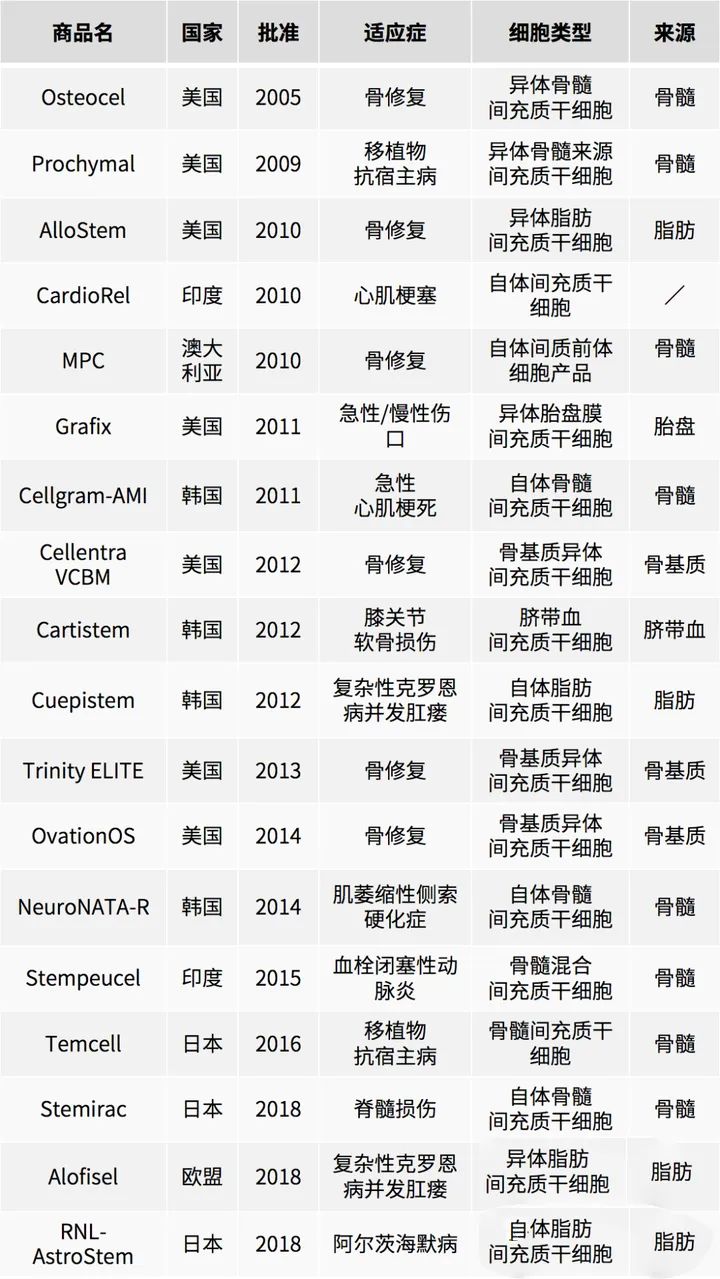
我国间充质干细胞药物临床试验默示许可汇总(截止至2024.1.17)
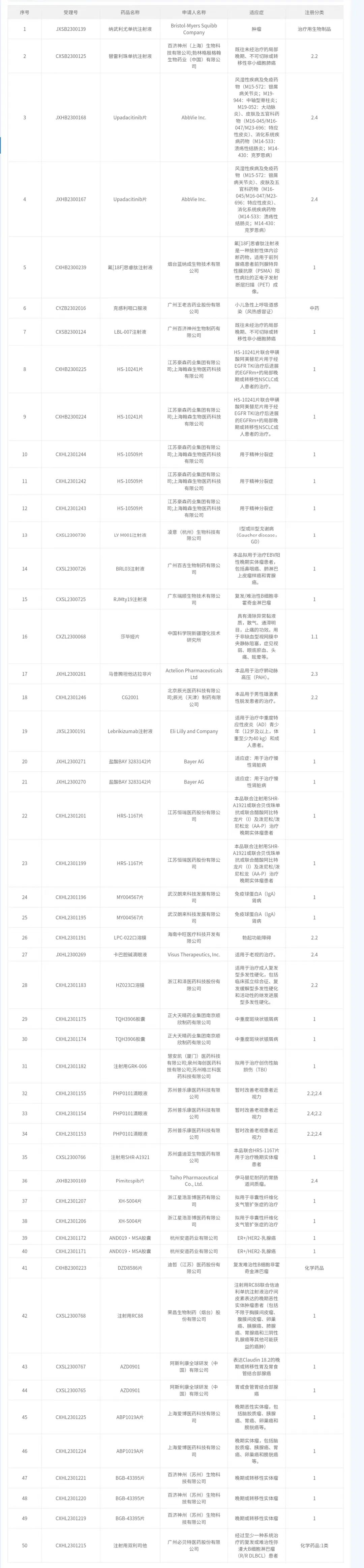
PART2 干细胞新药审批流程参考
新药的研发与上市是一个复杂的过程,除了临床前研究、临床试验(一期、二期、三期)这些关键步骤外,新药IND申请也是必不可少的环节。只有通过这些阶段,并且每一阶段都顺利通过审批,新药才能最终获得上市许可。
01新药临床研究申请
IND,全称为Investigational New Drug Application,是指干细胞产品通过临床前试验后向美国FDA(我国为“药监局”)提交的新药临床研究申请。如果在提交申请后30天内,FDA未驳回申请,那么该产品就可以进行临床试验。
02新药申请
在完成所有三个阶段的临床试验后,可以将研究资料和数据整理汇总,向FDA提交新药申请,即NDA。NDA的目的是确保上市药品的安全性、有效性和质量可控。一般情况下,FDA应在6个月的规定时间内完成NDA审评。但由于申请材料过多,大多数新药申请的审评时间会超过这个期限。
PART3干细胞监管政策
干细胞疗法作为一项新兴的技术,需要进行强有力的监管,在保证患者健康的同时,还有利于推动整个行业的健康发展。
2019年,我国发布了《关于做好2019年干细胞临床研究监督管理工作的通知》,明确了干细胞临床研究和应用规范采取“类双轨管理模式”,即由“国家药监局”和“卫健委”共同开展,以建立符合我国国情的干细胞技术规范。

2022至2023年,我国又相继颁布了《细胞治疗产品生产质量管理指南(试行)》、《“干细胞研究与器官修复”重点专项2022年度项目申报指南(征求意见稿)》、《人源性干细胞及其衍生细胞治疗产品临床试验技术指导原则(试行)》等多条法规,对干细胞产品的临床试验、质量管理、生产管理、临床应用(如干细胞与器官衰老、新型冠状病毒肺炎、心血管功能重塑)等方面,进行了更加规范的管理。

PART4 总结
2023年全球干细胞治疗市场收入为2.86亿美元左右,且具有广阔的发展潜力。预计到2028年,干细胞市场估值将高达6.15亿美元。目前全球范围内获批的干细胞产品主要集中在日本、韩国、美国、欧盟等地。近几年,我国也在大力扶持干细胞技术的发展,目前已将干细胞发展列入“十四五”发展规划。
相信在不久的将来,干细胞将会像“智能手机”一样,深入影响我们每个人的生活。我们也期待随着干细胞药物研究规模的不断扩大,再生医疗技术能让更多的患者获益!